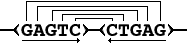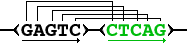Changes to previous online versions of reputer
We do not offer precalculated genomes any more. The online version of REPuter has
only little restrictions now (Our server capacity grows), so there is no reason to
offer any precompute genomes (which are mainly not needed). The current version of
REPuter offers a HTML 5 application for visualisation and filtering the results.
Textual Output: The result of a run can viewed/downloaded as a space
separated table. Optional the output can be filtered. The head of a sample output
looks like :
# 235 -3 8 reputer_bibitest_1091788224_479525172.xmlrpc
9 150 F 9 151 0 5.92e-02
8 150 F 8 152 0 2.37e-01
10 150 F 10 153 -1 4.44e-01
9 150 F 9 154 -1 1.60e+00
[1][2][3][4][5] [6] [7]
...
The first line, starting with '#' is a comment. The sequence length (235),
the maximum allowed distance ([-]3), the minimum repeat size (8) and the
processed file are described here. The following lines contain repeats found ,
one line each .
- repeat length of the first part
- starting position of the first part
- match direction
- repeat length of the second part
- starting position of the second part
- distance of this repeat
- calculated e-value of this repeat
Theoretical Background
This tool reports maximal forward, reverse, complemented, and reverse
complemented repeats for a given input sequence. The definition of 'maximality' as
in [1] basically limits the output to only the longest repeats in the sequence.
These may contain shorter repeats which are not explicitly reported.
Let your input sequence be a text string s of length
n. The characters in s are indexed from 0 to
n-1, therefore s can be written as
s=s
0s 1... s
n-1. For each reported repeat denoted by a triple ( l, i,
j), i.e. size, starting position of a piece of sequence and starting
position of its repeat counterpart, we postulate the size l>0
and the starting positions i, j ∈ [ 0, n-1].
REPuter distinguishes four different kinds of repeats:
- Maximal forward repeat, MFR
- Maximal reverse repeat, MRR
- Maximal complemented repeat, MCR
- Maximal palindromic (reverse complemented) repeat, MPR
The triple ( l, i, j) is a MFR if:
-
i ≠ j
(There is no identical starting position).
-
s is
i+1... s
i+l-1 =
sjs
j+1
... s
j+l-1
(Both parts of the repeat have the same size).
-
If 0 ≤
i-1, then s
i-1
≠
s
j-1
(If the first part of the repeat starts at a position greater or equal
to 0, then the characters immediately to the left of each part are
different).
-
If j+l ≤ n-1, then s i+l
≠ s j+l
(If the ending position of the second part of the repeat is less or
equal than the total input sequence size, then the characters immediately
to the right of each part are different).
[1] Gusfield, D., Algorithms on Strings, Trees, and Sequences, Cambridge
University Press, 1997
REPuter Sample Run
Consider the following 30 bases input sequence, which is a three-fold repetition
of 'gacagtcagt':
>5.seq
gacagtcagtgacagtcagtgacagtcagt
The REPuter engine produces the following raw data
output, starting with the input sequence name. Following, each line describes one
repeat, its size, starting position of the first part, one of the four possible
modes (F, P, R, C), then the starting position of the second
part.
The output below therefore reports two repeats, both starting at
position 0. The first part of the first repeat starts at position 0, its second part
at position 20.
# /tmp/5.seq.flat 30
20 0 F 20 10 0 2.30e-10
10 0 F 10 20 0 2.41e-04
Drawing the sequence in dark blue and the repeats in light blue this might look like this:

Note that according to the 'left character' rule 3. for MFRs in
the Theoretical Background section, we do not report a repeat like "10
0 F 10", since this short repeat will become part of "20 0 F
10".
Additionally, to keep the starting position information visible, each part of a
repeat is displayed on a separate strand: