ConCysFind is a pipeline tool searching conserved amino acids in protein sequences of plant kingdom
ConCysFind was developed on behalf of the department "Plant Biochemistry and
Physiology" at the University of Bielefeld. The development was supported by the
department "Computational Metagenomics". A pipeline developed by A. Sahm served
as a template, which searches for conserved cysteines of transcription factors
of the Plant Transcription Factor Database (PlantTFDB) and could previously
predict conservation of several transcription factors. In previous versions,
only conserved cysteines were considered. With ConCysFind searches for
conservation of other post translationally modified amino acids is possible:
tryptophan, serine, threonine, tyrosine and methionine. The search of conserved
amino acids is limited to the plant kingdom. For this purpose, 21 plant species
that represent high evolutionary diversity and are evenly spread among the
different plant taxa with consideration of one proxy species per species were
chosen. A phylogenetic tree of these species, based on the Tree of Life Web
Project (Maddison /et al/., 2007) can be viewed here:
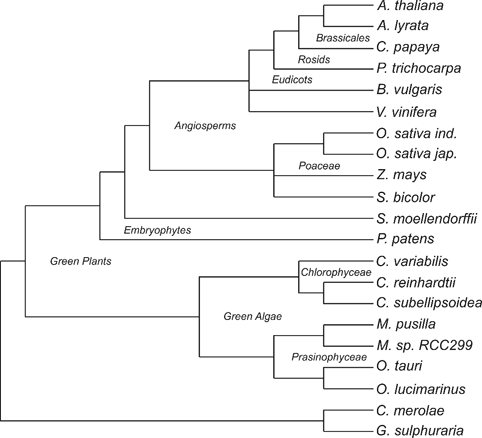 All protein sequences of these plant species were downloaded from
UniProt in FASTA format. This tool is working with this database.
All protein sequences of these plant species were downloaded from
UniProt in FASTA format. This tool is working with this database.
