With current sequencing technologies, it is feasible to generate a lot of
reads for a given genome. However, the assembly usually ends up in a set of contigs with
gaps between them. If the sequences of one (or several) related genome(s) are already
known, this information can be used to estimate the order and orientation of the contigs
towards each other. This helps in the finishing phase of a sequencing project since it
eases the design of specific primer sequences to fill the gaps.
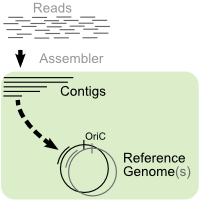
Description
This is a toolsuite for ordering and orienting contigs in a comparative genomic
fashion. Currently two programs are included:

|
r2cat, the related reference based contig
arrangement tool can be used to order a set of contigs with respect to a single
reference genome. This is done by mapping the contigs onto the reference using a
q-gram filter. The mapping is visualized in a synteny plot. |
|
treecat - phylogenetic tree based contig
arrangement tool takes several genomes and their relationships in a phylogenetic
tree into account to estimate a possible ordering of the contigs. |
Sign on to this mailinglist if you want to be up to date about bugfixes and latest
developments for r2cat and treecat.
Starting the application It is easy to start the programs of this
toolsuite. All that is needed is Java WebStart. To try out an application select a r2cat or
treecat in navigation above. Other tools Here is a list of other tools
which deal with the same problem of arranging contigs:
|
