Abstract
Gene regulation is the process through which an organism effects spatial and temporal
differences in gene expression levels. Knowledge of cis-regulatory elements as key
players in gene regulation is indispensable for the understanding of the latter and
of the development of organisms. We here present the tool jPREdictor for the fast
and versatile prediction of cis-regulatory elements on a genome-wide scale. The
prediction is based on clusters of individual motifs and arbitrary combinations of
these into multi-motifs with selectable minimal and maximal distances. Individual
motifs can be of heterogenous classes, such as simple sequence motifs or position-specific
scoring matrices. Cluster scores are weighted occurences of multi-motifs, where the
weights are derived from positive and negative training sets. The jPREdictor was used
for the prediction of Polycomb/Trithorax Response Elements in Drosophila melanogaster,
where the number of predictions are doubled compared to Ringrose et al. (2003), Dev
Cell 5, 759-771, at the same level of specificity.
Tasks to perform with the jPREdictor
The jPREdictor can handle and perform several tasks. All of them were used step-by-step
in the prediction of special cis-regulatory elements called PREs/TREs in Drosophila
melanogaster [Fiedler and Rehmsmeier, 2006]. In short, a prediction uses weighted motifs
to score a section of the genome and if the score exceeds a threshold a regulatory
element was found.
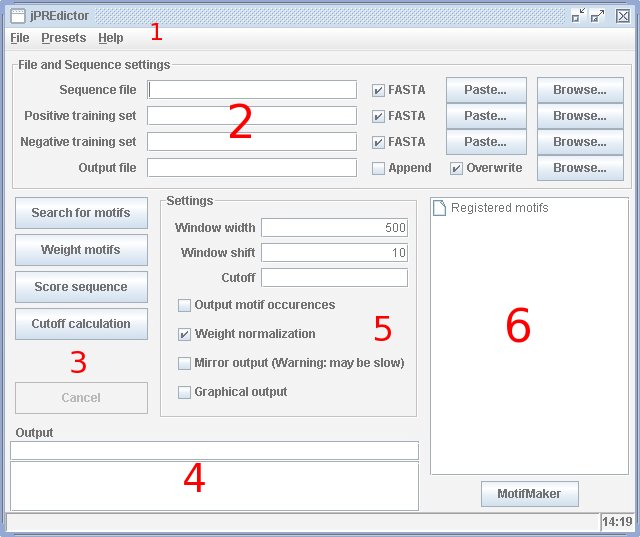
The first task the jPREdictor must be able to do is to properly describe, to define and
to re-read motifs. Motifs in this sense are nucleotide or amino-acid sequence patterns
that are widespread and have, or are conjectured to have, a biological significance [
Wikipedia]. With the jPREdictor motifs can be defined by using the build-in MotifMaker,
the command-line parameter -d or the option file. In the programs main window [see picture]
the motifs used for all further tasks are shown in section 6. Pressing the button "MotifMaker"
opens the motif maker dialog where the motifs can be changed. The menu "Presets"
(section 1 in the picture) provides the user with two motif sets used previously for the
prediction of PRE/TREs [Ringrose et al, 2003; Fiedler and Rehmsmeier, 2006].
Once the motifs are known and properly represented the jPREdictor can apply each motif a weight. This is done by pressing the "Weight motifs" button in section 3. The program than searches the motifs on the sequences of two training sets. The first training set, the model or positive training set, represents the regulatory elements, which are to be predicted with the program. Therefore, the model contains sequences which are such regulatory elements. The second set, called background or negative training set, contains sequences which are not regulatory elements or from which the sequences to be predicted should be distinguishable. This can be randomly generated sequences or special parts of the genome. Both training sets can be given to the program in section 2, e.g. use the "Browse"-buttons to give files. Another way is to load an option file where the motifs and corresponding training sets are specified (menu "File" in section 1, than "Load..."). In the case of the PRE/TRE prediction the model contained known PRE/TREs and the background contained promotor sequences from heatshock and other genes [Ringrose et al, 2003]. The motifs weight itself is a log-odd-score and calculated as the logarithm of the following fraction: number of the motif found in the model divided by the number in the background. Some normalization is done, too, either by sequence number (should be used if all sequences are of equal length) or by sequence' lengths.
The weighted motifs are then used to score genomic sequences (press "Score sequence" in section 3). The sequence file must be given in section 2 or uploaded together with the option file. The jPREdictor performs the scoring by shifting a sequence window of defined width, e.g. 500 nucleotides, over the genome. Within the window motifs are searched and the corresponding weights are summed up. Normally, high scoring sequence windows are meant to be regulatory elements, whereas low scoring windows have no meaning, they are noise (background). The scores for the sequence can be examinded in the graphical score plot browser (check button "Graphical output" in section 5). This is only recommended for short sequences. When scoring sequences the output is normally a file (to be specified in section 2) with the score for every sequence window. Specifiing a cutoff (section 5) activates automatic comprising to bands. The output is than the start and end of the band on the genomic sequence together with minimum, maximum and mean score.
But what minimal score is required to distinguish looked-for regulatory elements from genomic noise, what score is neccessary to separate the wheat from the chaff? To provide at least a consideration the built-in cutoff calculator was developed. This tool scores randomly generated sequences and tables all found scores. From the table cutoffs can be obtained, which correspond to E-values, the expected number of higher-scoring sequence windows for this score.
Motif maker explained
The motif maker was developed to graphically display motifs. A list of defined motifs is shown in section 1 [see picture]. This is the same list as in the jPREdictor main window and contains all motifs to be used for weighting and scoring.
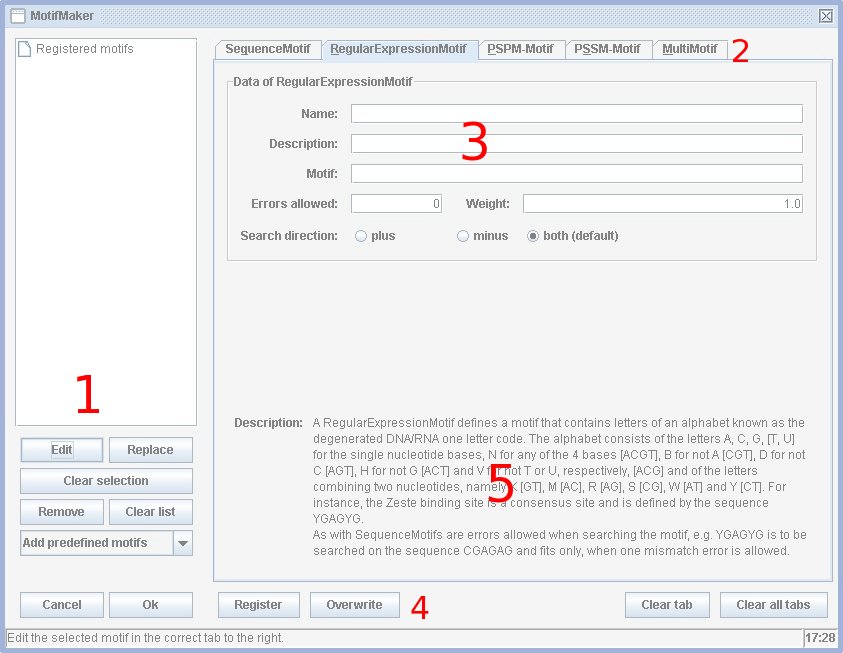
The jPREdictor understands four types of primitive motifs, SequenceMotifs, RegularExpressionMotifs, PSPMs and PSSMs and one complex motif type, MultiMotifs. The motif types are shown and are to be selected in section 2. If the "Edit"-button (section 1) is pressed with a selected motif from the list the corresponding motif type is displayed and the fields within (section 3) are filled with the motifs information like name, description, sequence, weight, ...
SequenceMotifs are simple nucleotide motifs and are composed from the letters of the DNA alphabet ('A', 'C', 'G', 'T'), e.g. DSP1 has the sequence GAAAA. RegularExpressionMotifs are nucleotide motifs, too, but use the extended IUB DNA alphabet, which is 'A', 'B' (not 'A'), 'C', 'D' (not 'C'), 'G', 'H' (not 'G'), 'K' ('G' or 'T'), 'M' ('A' or 'C'), 'N' (all), 'R' ('A' or 'G'), 'S' ('C' or 'G'), 'T', 'U', 'V' (not 'T' or 'U', respectively), 'W' ('A' or 'T') and 'Y' ('C' or 'T'). PSPM and PSSM are table motifs. PSPM means position specific probability matrix, for every position in the motif the nucleotides have specific occurence probabilities. For PSSMs (position specific score matrix) such probabilties are rather log-odd-scores, calculated from the occurence probabilities against a background nucleotide composition.
MultiMotifs are the complex motif type within the jPREdictor. They are a composition of two or more arbitrary motifs, simple as well as complex ones, together with distance informations. Distances are measured from the end of one motif to the beginning of the next. The sequence GAAAAGAAAA contains two times the motif DSP1 (sequence GAAAA) and the distance is zero, because no other nucleotide occurs between them. For overlapping motifs negative distances are possible. Example: Dejardin et al, 2005, always found the DSP1 motif near the PHO motif (core sequence is GCCAT) to built up a functional unit, the MultiMotif would be represented as PHO-(0,40)-DSP1. For the prediction of PRE/TREs Ringrose et al, 2003, have shown, that double motifs are much more practical to distinguish between regulatory and non-regulatory elements. Thus, it is strongly recommended to use DoubleMotifs for the prediction of regulatory elements. To do so, first define and register (button "Register" in section 4) all simple motifs, then click on MultiMotif (section 2) and move all motifs to the "Motifs to combine"-list. Then press the button "Build double motifs". Don't forget to remove the simple motifs from the list to the right, they are no longer needed. In the main window it is possible to save the motif list to an option file (menu "File", then "Save...") for repeated use.
The cutoff calculator explained
The cutoff calculator was developed to provide the user with an idea of how high the scores can become by chance when using the given motif set. To perform this task the cutoff calculator generates random DNA sequences and scores them using the weighted motifs. The scoring function is the same as in the main program: the motifs are searched in a sequence window of defined width, the weights are added up to a score, but this time the program counts how often a score was found. Note, that this description is short of the truth. To fully understand how it works, the understanding of bands is neccessary. A band comprises sequence windows, which are above the cutoff and overlap each other. A band is always maximal in width and no two bands overlap for the same cutoff. The cutoff calculator now does not simple count the scores of the sequence windows but does count bands. It is imaginable, that their exists a low cutoff where the whole sequence is one band, and that their exists a high cutoff where no band exists any more. These two extreme values mark the minimum and maximum cutoff.
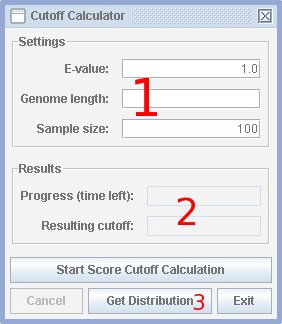
Example: The genome size may be 110 million nucleotides (almost the size of the Drosophila melanogaster genome). The sample size may be 100, which is appropriate for an E-value of 1. The calculator now generates step-by-step 100 random sequences of size 110 million (11 billion nucleotides overall), scores it and calculates for every reasonably cutoff the number of bands. The cutoff for an E-value of 1 is the one where the number of bands equals the sample size. This is the cutoff for which only one false positive band can be expected, when the genome is scored.
In section 1 [see picture] the genome length can be spezified. Note, that an E-value is only valid together with a sequence length. If the E-value is changed the number of samples change, too. Lesser E-values require more samples. One sample is the size of the genome. In section 2 the progress meter is shown, it gives the percent done and the estimated time to finish calculation. When the cutoff calculation has finally stoped the resulting cutoff for the specified E-value is given. Section 3 shows the "Get Distribution" button, where the nucleotide distribution of the randomly generated sequences can be changed. Pressing the button results in opening the sequence file given in the main window and obtaining the nucleotide distribution from the DNA sequences within the file. If no sequence file is given, an error dialog pops up and the currently used distribution is shown. The distribution can directly be specified by changing the global distribution. Use the option file directive global_background for that purpose.
Comand line example
Option file samples
The first sample defines the long GAF motif, which is searched with one mismatch error allowed and only the normal direction on a sequence and not reversed/complementary.
[Motif]
name = G10
description = the long GAF binding site of size 10
motif = SEQUENCE
GAGAGAGAGA
errorNumberAllowedForMatch = 1
searchDirection = normal
The next sample defines a PHO motif from multiple short sequences found in some literature. The empty lines and the spaces within the short sequences are ignored. The motif is first calculated as a PSPM (position specific probability matrix), i.e. for every position the nucleotides have occurence probabilities. By giving a background, the PSPM is recalculated to a PSSM (position specific score matrix) with a score (log-odd-score) for every nucleotide in every position. If such a PSSM motif is searched the sequence-character-corresponding score from every positions is added and, if the sum-score exceeds the threshold, the motif was found.
[Motif]
name = PHO
description = PHO core with leading and trailing parts
motif = MULTI_SEQUENCE
GGCAG CCAT TTTCC
CGCAG CCAT TTTCC
GTCGG CCAT TAAAA
GGAAG CCAT AACGG
background =
0.287 0.213 0.213 0.287
threshold = 10.2
The next sample defines a triple motif (MultiMotif) of three single motifs. The first two motifs must stick close together (maximal distance is 30), the third, G10, can occur with greater distances.
[MultiMotif]
name = PHO-G10-G10
first_motif=PHO
next_motif=0,30,G10
next_motif=0,200,G10
The last sample defines the 6 double motifs PHO-G10:PHO-G10, PHO-G10:G10, PHO-G10:PHO, G10:G10, G10:PHO, PHO:PHO. Motifs are identified for their name. The distance between the two motifs in every generated DoubleMotif is 0 to 219, messured from the end of one to the beginning of the next motif. Note, that PHO-G10 seams to be a DoubleMotif itself, thus, the first resulting MultiMotif PHO-G10:PHO-G10 is in fact a QuadrupleMotif and all compositions with this motif are TripleMotifs.
[MultiMotifList]
distance=0,219
PHO-G10, G10, PHO
Some notes about searching MultiMotifs
MultiMotifs are searched the way that first the corresponding single motifs are searched. From the occurances of the single motifs it is decided, whether a MultiMotif occured by taking the distance information into account. The distance between two motifs is always messured from the end of the first to the start of the latter motif. Due to this the motifs cannot be found overlapped iff the distances are non-negative.
Another thing to consider is the order of the motifs. The DoubleMotif A-B is always also searched reversed, thus occurences of B-A are found automatically, too. Another thing are triple motifs and MultiMotifs of higher order. MultiMotifs are also searched reversed, but not permutated. Thus, for the triple motif A-B-C occurences of C-B-A are found, but not for instance B-A-C. But there might be the case when one motif occurs often close to another without considering their order (satellite). When doubling motifs this pair should be defined first:
[MultiMotif]
name=ASat
firstmotif=A
secondmotif=0,30,Satellite
and afterwards used for doubling:
[MultiMotifList]
distance=0,219
A,ASat,B,C
This MultiMotifList section yields the DoubleMotifs A-A, A-ASat, A-B, A-C, ASat-ASat, ASat-B, ASat-C, B-B, B-C, C-C. Note, that every DoubleMotif containing a satellite is in fact either a QuadrupleMotif or a TripleMotif, but not flat. The advantage of having non-flat MultiMotifs is, that more permutations of the motifs in it are possible, e.g. every motif pair with a satellite does not only occur in two permutations A-B and B-A; but for instance ASat-B can occur as A-Sat-B, Sat-A-B, B-A-Sat or B-Sat-A, but not as A-B-Sat, Sat-B-A.
Install Java
For running the jPREdictor you need in a functional Java in version 1.5 or higher installed on your computer.
If no Java (or an outdated version) is installed on your computer, please download and install the newest version of
the Java Runtime Environment. First, click here and you will be directed to the Java Download Site. Then click "Download Now"
and follow the Step-By-Step Java installation guide. If you have problems to install Java on your computer
(maybe you do not have the permissions) please contact your administrator.
Appendix A: Command Line Parameter
To get a list of supported command line parameter type
java -jar jPREdictor.jar -?
on command line.
Appendix B: Special Command Line Parameters
The special purposes command line parameters and can be received on command line by
java -jar jPREdictor.jar -hs .
Appendix C: Settings for the option file
This help can be received by typing java -jar jPREdictor.jar -ho on
the command line. Only one option file gan be given to the jPREdictor program. Note, that the
option file is evaluated always subsequent to all other parameters. This is importent,
because some options in the file overwrite parameters, e.g., you give a file 'sequ'
using parameter -f and a option file 'optionfile' using parameter -o. The complete command line call is then:
java -jar jPREdictor.jar -o optionfile -f sequ
The parameter -f is overwritten by the line sequence_filename= in the option file.
