|
KnotInFrame - a -1 PRF prediction pipeline:
The -1 PRF prediction pipeline KnotInFrame is composed of three consecutive steps:
1. In the search phase, we scan the input sequences for occurrences of the consensus slippery site in the correct reading frame.The downstream region of each slippery site is checked for suitability as a frameshift signal. This is done by comparing the minimal free energy of an enforced pseudo-knotted folding with the minimal free energy of a freely folded structure. The former energy is computed by a special version of pKiss, the latter by an RNAfold-like program.
2. In the filtering phase, three criteria based on the energy values of the free and the constrained folding are applied to reduce the number of candidates.
3. In the ranking phase, the candidates passing all filters are ranked by an evaluation function based on the normalized dominance of the pseudoknot.
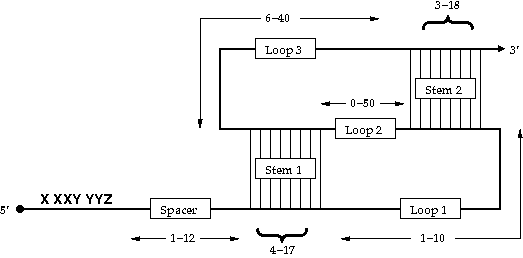
Figure 1: The consensus -PRF signal derived from the RECODE database.
The slippery sequence has the consensus X XXY YYZ, where XXX stands for any three identical nucleotides, YYY for either three As or three T/Us and Z for any nucleotide. The spacer region must contain at least 1 nt and not more than 12 nt.
KnotInFrame
In-/Output values
INPUT :: DNA/RNA sequences
The input file should contain at least one DNA or RNA sequence in FASTA format.
OUTPUT :: DNA/RNA structures
The following image shows the output of the example call
knotinframe < example.mfa
Colored elements are not part of the output. Click on the image for a larger view.
| >pol_m_vir_mhv|AF029248.1 |
| |
| Rank: | 1 | |
| Slippery sequence: | AAAAAAG |
| Slippery position: | 56 |
| Substring length: | 60 |
| Deltarel: | 0.070 |
| 63 | AAAGGTAAGACCAAGCATGGTAGGGGTAGAGTCAGACGTAACCTTAGAAAAGGCGTGAAA | 123 |
| -10.40 | .[[[[[...((((....))))..((......))..{{{{.]]]]].......}}}}.... | knotted structure |
| -6.20 | ...........((.((....(((((................)))))......)).))... | nested structure |
| |
| >pol_m_vir_eiav |
| |
| Rank: | 1 |
| Slippery sequence: | AAAAAAG |
| Slippery position: | 1 |
| Substring length: | 60 |
| Deltarel: | 0.038 |
| 8 | GGGAAGCAAGGGGCTCAAGGGAGGCCCCAGAAACAAACTTTCCCGATACAACAGAAGAGT | 68 |
| -17.40 | .........[[[[[[...{{{{]]]]]]............}}}}................ | knotted structure |
| -15.10 | ((((((...(((((((....)).)))))..........))))))................ | nested structure |
|
|
| |
| |
| Rank: | 2 |
| Slippery sequence: | AAAAAAG |
| Slippery position: | 136 |
| Substring length: | 120 |
| Deltarel: | 0.002 |
| 143 | GAATACAATGTCAAGGAGAAGGATCAAGTAGAGGATCTCAACCTGGACAGTTTGTGGGAGTAACATATAATCTAGAGAAAAGGCCTACTACAATAGTATTAATTAATGATACTCCCTTAA | 263 |
| -24.80 | .[[[[[..((((.(((....(((((........)))))...))).))))......{{{{{{{..........(((((.......)).))).....]]]]].........}}}}}}}.... | knotted structure |
| -24.60 | ....((((((((.(((....(((((........)))))...))).))))..))))(((((((.(((.(((((((......((.....))....))).))))....))).))))))).... | nested structure |
|
The example consists of two sequences, thus you get two sequence results (magenta boxes).
They start with a identification line, followed by a number of candidate blocks (blue boxes).
The first character of the identification line is the FASTA typical >, followed by the name of the sequence.
Every two sequence results are separated by an empty line.
The number of candidate blocks is limited by the parameter numberOutputs.
Candidate blocks have a general indent of two white-spaces.
Each candidate block has two parts. The info header and the structure body.
Within the info header each information provides a key and a value, separated by a colon and a number of white-spaces.
The structure body keeps three lines, which are further divided by two white-spaces into three parts. First line gives start position, sub-sequence and end-position of the predicted site, containing the pseudoknot. The slippery sequence is directly upstream located.
Last two lines have three fields as well. First field gives the free energy of the prediction in kcal/mol. Second field is the prediction itself in Vienna dot bracket representation. Last field indicates the type of prediction (either "knotted structure" or "nested structure").
As sequence results, candidate blocks are separated by white-spaces. Should the number of available candidate blocks exceeds numberOutputs the warning "Warning: some further results are not shown. Increase --numberOutputs to display them!" is issued.
Parameter
| temperature |
The energy parameters used in the calculation have been measured at 37 C. Parameters at other temperatures can be extrapolated, but for temperatures far from 37 C results will be increasingly unreliable.
|
| thermodynamic model parameters |
Read energy parameters from a file, instead of using the default parameter set. See the RNAlib (
Vienna RNA package) documentation for details on the file format.
Default are parameters released by the Turner group in 2004 (see [
mat:dis:chil:schro:zuk:tur:2004] and [
tur:mat:2010]). A visit of the aforementioned author's
Nearest Neighbor Database might also be informative.
|
| windowSize |
If a slippery site is detected a substring downstream is analysed. --windowSize sets the maximal length of this substring, which is 120 by default. Must be a multiple of --windowIncrement.
|
| windowIncrement |
The --windowSize bp long substring, downstream to the slippery site, is analysed in different chunks sizes. These chunks grow with --windowIncrement bp. Maximal size is --windowSize. Default value is 20.
|
| minKnottedEnergy |
The pseudoknot structure induces the ribosomal frameshift, thus it should have a stability of at least -7.4 kcal/mol.
|
| minEnergyDifference |
The candidate subsequence should be more likely fold a pseudoknot than a nested structure. Thus, candidates where the nested energy + --minEnergyDifference < knotted energy, are ruled out. The default value is -8.71 kcal/mol.
|
| numberOutputs |
Some sequences have a high amount of possible slippery site candidates, thus output is cut off after printing the best --numberOutputs results, which is 10 by default.
|
Recommended Input:
Short sequences up to 120 nucleotides can be folded within seconds. Once a sequence is folded, shorter substrings can simply be backtraced. The expected run time for an average sized bacterial genome of 5MB is 1.5 hours. In practice, a filter already discards a significant amount of slippery sites, before the expensive folding step, which decreases the runtime. Thus the KnotInFrame-Tool requires about 4.5 CPU-hours to analyse the 8.76 MB coding sequence of yeast.
|
