

Next: Format of Textual Output
Up: Efficient Multiple Genome Alignment
Previous: Scripts
Output
The results of mga are written to a number of files whose prefix can
be specified by option -o. The following files may be generated:
-
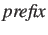 .summary
.summary
- This file contains the parameters used during the computation, a
description of the sequences and a short statistics on the number
and length of the various regions.
-
.align
- This file contains the data of the alignment. By default, only the
positions of all matches, aligned and unaligned gaps are given.
This results in a short output which is useful for getting a brief
overview of the structure of the alignment. The actual sequence
data of the matches and aligned gaps are presented if requested
using options -match / -bl and -clustalw
/ -msascript / -greedy / -xdrop.
-
.reg
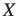
- These files contain the sequence data of all aligned regions and
matches in FASTA format. There will be a file for every continous
region number X. The files will only be created if option
-alignedseqs is specified.
-
.gap.seq
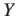
- These files contain the sequence data of all unaligned gaps in FASTA
format. There will be a file for every gap number X and every
sequence number Y. The files will only be created if option
-gap is specified.
-
.chain
- This file contains all chained matches and is generated by
option -chain. The file can, for example, be parsed by
a graphical user interface that visualizes the backbone of anchors
and computes alignments of short gaps on demand.
-
.xml
- This file contains all information in a structured way using
XML-format. It can be used to postprocess the output of mga, e.g.,
to generate HTML pages containing the alignment. This is
demonstrated by mga2html.pl which is included in the
distribution.
If option -o is not specified the output will be written to
stdout. This does not work for all options, e.g., option
-gap requires option -o to be present.
Next: Format of Textual Output
Up: Efficient Multiple Genome Alignment
Previous: Scripts
Jan Krueger
2012-12-14